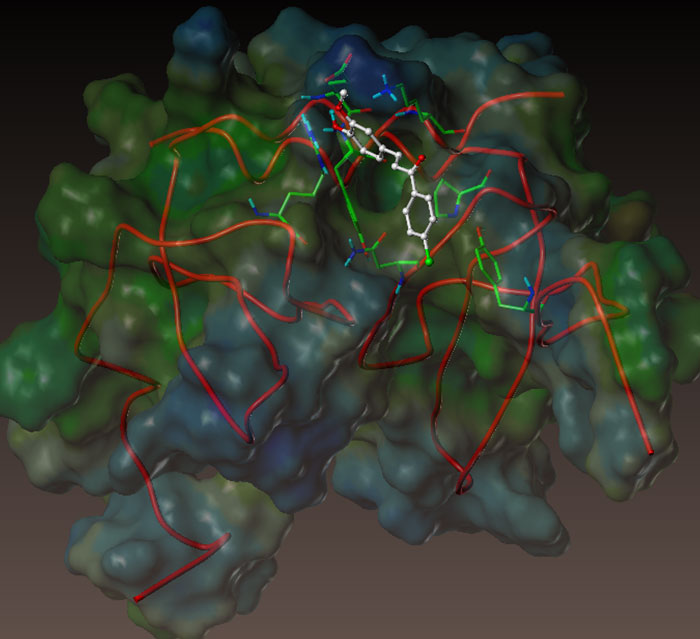
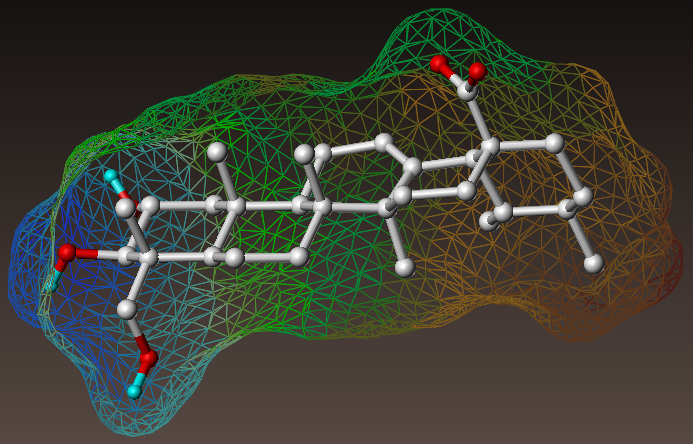
The discovery of active compounds includes different phases of R&D. Greenpharma accompanies customers in the early research stage by proposing innovative services in the library design and the selection of molecules for biological evaluations.
In this end, we use our chemoinformatic platform and the Ambinter & GPDB database as sources for compounds. Depending on the needs and the level of information, the selection may be performed according to structural diversity or similarity. We can also do virtual screening.
Furthermore, we propose tools for drug repurposing such as SELNERGYTM
Last, ASQR/QSPR approaches are useful for the predictions of biological activities or physico-chemical properties.